I-cel (ziekte)
Niet te verwarren met I-cel (darm).
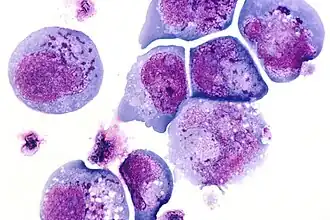
I-cellen zijn cellen met een groot aantal donkere insluitsels in het cytoplasma en de kern van de cel. Het zijn aggregaten van kleurbare stoffen, meestal eiwitten.[1] Deze metabolisch inactieve aggregaten zijn niet omgeven door een membraan en bestaan uit vetten, eiwitten, koolhydraten, pigmenten en uitscheidingsproducten. I-cellen worden geassocieerd met neurodegeneratieve ziekten. Ze worden gezien bij mucolipidose II en mucolipidose III, ook wel inclusiecel- of I-celziekte genoemd, waarbij het lysosomale enzymtransport en -opslag is aangetast.
Insluitsels werden voor het eerst beschreven in de late 19e en 20e eeuw. Een van de vroegste onderzoekers die in verband wordt gebracht met de ontdekking van insluitsels is Fritz Heinrich Jakob Lewy. Hij ontdekte bijzondere insluitsels in zenuwcellen van bepaalde hersenkernen bij patiënten met paralysis agitans (ziekte van Parkinson), die later door Gonzalo Rodriguez Lafora een "Lewy Body" zouden worden genoemd.[2] Deze ontdekking is een van de beroemdste vroege observaties van insluitsels.
Oorzaken en erfelijkheid
Bij I-celziekte ontstaan de insluitsels door een defect in het transport van enzymen naar de lysosomen, waar afvalstoffen worden afgebroken. Dit defect wordt veroorzaakt door een mutatie in het GNPTAB-gen in het enzym N-acetylglucosamine-1-fosfotransferase.[3] Dit leidt ertoe dat de lysosomale enzymen niet met mannose-6-fosfaat kunnen worden gelabeld. Zonder dit label kunnen de enzymen niet correct aan de lysosomen worden geleverd en worden afvalstoffen opgeslagen als insluitsels in plaats van afgebroken. Deze insluitsels verstoren celfuncties en veroorzaken symptomen zoals ontwikkelingsachterstanden, abnormale groei, grove gelaatstrekken en vergrote organen. Deze mutatie wordt autosomaal recessief overgeërfd, dus beide ouders moeten drager zijn van één kopie van het gemuteerde gen om deze aandoening bij verwanten te kunnen ontwikkelen.[4]
Klinische betekenis
I-celziekte gaat gepaard met verschillende klinische kenmerken die het uiterlijk, de orgaanfunctie en de groei beïnvloeden. De ernst van deze symptomen verschilt per persoon, hoewel de prognose slecht is vanwege het systemische karakter van de ziekte. Patiënten met I-celziekte kunnen ook een verminderde cognitieve en motorische ontwikkeling ervaren. Personen kunnen ook grove gelaatstrekken hebben, zoals een prominent voorhoofd, een platte neusbrug of een verdikte huid.[3]
Skeletafwijkingen zoals dysostosis multiplex of een kleine gestalte komen ook vaak voor. De orgaanfunctie kan worden beïnvloed door hepatosplenomegalie en vergroting van de lever ((hepatomegalie) en milt (splenomegalie), of hartproblemen zoals klepafwijkingen. De ziekte kan zich neurologisch manifesteren in cognitieve stoornissen of epileptische aanvallen, of in gewrichts- en ledemaatproblemen zoals artropathie, progressieve artritis en stijfheid.
Andere mogelijke symptomen van de ziekte zijn maag-darmproblemen, problemen met het gezichtsvermogen of gehoor, immunologische problemen (verhoogd infectierisico) en een kortere levensverwachting. Mucolipidose II is ernstiger dan mucolipidose III en resulteert doorgaans in de dood van de patiënt binnen de eerste tien levensjaren.[5]
Diagnose
.jpg)
Om I-celziekte te diagnosticeren, voeren specialisten een combinatie uit van klinische evaluaties, biochemische tests en genetische analyses. Eerst bekijken artsen de medische voorgeschiedenis van de patiënt op symptomen zoals groeiachterstand, grove gelaatstrekken, orgaanvergroting of skeletafwijkingen. Deze eerste onderzoeken onthullen vaak kenmerken zoals hepatosplenomegalie, gewrichtsstijfheid of dysostosis multiplex.[6]
De volgende methode om de ziekte te diagnosticeren, is biochemische tests overwegen, zoals het meten van de activiteit van de lysosomale enzymen in bloed of urine. Bij personen met de ziekte verschijnen specifieke enzymen (zoals β-glucuronidase en N-acetylgalactosamine-4-sulfatase) in hogere concentraties met verminderde activiteit (als gevolg van geen lokalisatie). Urineonderzoek kan ook verhoogde niveaus van glycosaminoglycanen (GAG's) aantonen, complexe koolhydraten die zich ophopen bij lysosomale stapelingsziekten. Daarnaast kunnen specifieke enzymtesten worden gebruikt om de lysosomale enzymactiviteit te bepalen.[6]
Als de biochemische tests lysosomale enzymactiviteit aantonen die wijst op I-celziekte, voeren specialisten genetische tests uit om GNPTAB-genmutaties te identificeren. Om specifieke mutaties te identificeren, kunnen artsen Sanger-sequencing of next-generation sequencing gebruiken. Familieleden kunnen ook genetische tests ondergaan om dragerschap vast te stellen. Röntgenfoto's en echo's kunnen ook worden gebruikt om skelet- of orgaanafwijkingen te evalueren, en MRI- of CT-scans kunnen worden gebruikt om de hersenstructuur te onderzoeken bij personen met neurologische symptomen. Soms kan een biopsie van aangetast weefsel insluitlichaampjes in de cellen aan het licht brengen.
Een juiste diagnose is gericht op het onderscheiden van I-celziekte van andere lysosomale stapelingsziekten, wat moeilijk blijkt vanwege hun vergelijkbare klinische kenmerken. Identificatie van specifieke enzymdeficiënties en genetische tests helpen bij het stellen van de juiste diagnose. Deze diagnoses zijn essentieel voor het behandelen van de symptomen van de ziekte, hoewel er geen genezing bekend is.
Behandeling
Omdat er geen genezing is voor I-celziekte, blijft de behandeling zich richten op ondersteunende zorg en behandelstrategieën om de symptomen te verlichten en de kwaliteit van leven te verbeteren. Een multidisciplinaire aanpak is noodzakelijk voor de behandeling van de ziekte vanwege de meervoudige systeemstructuur. Over het algemeen is een team van zorgprofessionals betrokken, waaronder genetici, neurologen, orthopedisch specialisten en fysiotherapeuten.[7]
Fysiotherapie en ergotherapie worden gebruikt om mobiliteitsproblemen aan te pakken. Deze therapieën helpen bij het beheersen van gewrichtsstijfheid, het bevorderen van de motoriek en het vergroten van de spierkracht. Soms zijn chirurgische ingrepen nodig om skeletafwijkingen te herstellen of gewrichtspijn te verlichten. Voedingsondersteuning wordt ook gebruikt om voedingsproblemen of groeiachterstanden bij getroffen personen te bestrijden. Deze voedingsondersteuning maakt specifieke dieetplannen of het gebruik van voedingssondes mogelijk.[8]
Regelmatige behandeling van complicaties zoals hart- of ademhalingsproblemen is cruciaal. Naast fysiotherapie wordt counseling ingezet voor getroffen personen en families om emotionele steun te bieden. Een studie die de resultaten van hematopoëtische stamceltransplantatie bij patiënten met mucolipidose II onderzocht, toonde aan dat na hematopoëtische stamceltransplantatie de ruwheid van de huid van de patiënt aanzienlijk verbeterde, de spierspanning in de ledematen aanzienlijk afnam en de grove en fijne motoriek verbeterde.[3]
- Dit artikel of een eerdere versie ervan is een (gedeeltelijke) vertaling van het artikel I-cell op de Engelstalige Wikipedia, dat onder de licentie Creative Commons Naamsvermelding/Gelijk delen valt. Zie de bewerkingsgeschiedenis aldaar.
- ↑ Ramón, Ana, Señorale-Pose, Mario, Marín, Mónica (2014). Inclusion bodies: not that bad…. Frontiers in Microbiology 5. PMID 24592259. PMC 3924032. DOI: 10.3389/fmicb.2014.00056.
- ↑ Engelhardt, Eliasz (October 2017). Lafora and Trétiakoff: the naming of the inclusion bodies discovered by Lewy. Arquivos de Neuro-Psiquiatria 75 (10): 751–753. PMID 29166468. DOI: 10.1590/0004-282X20170116.
- ↑ a b c He, Si-jia, Li, Dong-jun, Lv, Wen-qiong, Tang, Wen-hao, Sun, Shu-wen (6 July 2023). Outcomes after HSCT for mucolipidosis II (I-cell disease) caused by novel compound heterozygous GNPTAB mutations. Frontiers in Pediatrics 11. PMID 37484777. PMC 10359890. DOI: 10.3389/fped.2023.1199489.
- ↑ National Library of Medicine. (2015, May). Mucolipidosis II alpha/beta: Medlineplus genetics. MedlinePlus. https://medlineplus.gov/genetics/condition/mucolipidosis-ii-alpha-beta/#inheritance
- ↑ Wang, Yu, Ye, Jun, Qiu, Wen-juan, Han, Lian-shu, Gao, Xiao-lan (February 2019). Identification of predominant GNPTAB gene mutations in Eastern Chinese patients with mucolipidosis II/III and a prenatal diagnosis of mucolipidosis II. Acta Pharmacologica Sinica 40 (2): 279–287. PMID 29872134. PMC 6329779. DOI: 10.1038/s41401-018-0023-9.
- ↑ a b Khan, Shaukat A., Tomatsu, Saori C. (17 september 2020). Mucolipidoses Overview: Past, Present, and Future. International Journal of Molecular Sciences 21 (18): 6812. PMID 32957425. PMC 7555117. DOI: 10.3390/ijms21186812.
- ↑ NORD. (2023a, November 20). Mucopolysaccharidoses - symptoms, causes, treatment: Nord. National Organization for Rare Disorders. https://rarediseases.org/rare-diseases/mucopolysaccharidoses/
- ↑ Khan, Shaukat A., Tomatsu, Saori C. (17 september 2020). Mucolipidoses Overview: Past, Present, and Future. International Journal of Molecular Sciences 21 (18): 6812. PMID 32957425. PMC 7555117. DOI: 10.3390/ijms21186812.